【重要】有机化学计算工具箱
感觉课题那边写的东西越来越多,已经超过这边的内容了,这边就改为工具箱,讲些软件相关的基础吧。
软件基础?
安装与调整
需要安装的包括:服务器相关的上传和命令软件和化学结构绘制查看软件。
SFTP软件与SSH软件
SFTP
WinSCP/Xftp免费许可证版/Filezilla
SSH
Putty/XShell免费许可证版
XShell和Xftp免费版的获取可以参考:
https://www.xshell.com/zh/free-for-home-school/
(现在可以直接在上述页面下载对应版本)
SFTP
SSH
代码编辑器
Notepad++(Notepad++因为程序员政治立场不建议,但是我们组都是在用这个的……反正我不用)
化学式绘制软件
ChemOffice 20~22 for Windows / ChemDraw 20~22 for Mac 一键破解
参考自:https://github.com/Z-H-Sun/CS_CCME_Posts/blob/hidden/cos/cos20.md
Windows补丁地址:
谷歌:https://drive.google.com/open?id=1FT3l0msiLPsarmT78JQ8_sgbdJLMY4_X
百度:https://pan.baidu.com/s/1smDXW8GNTPErq_yjilDhLg?pwd=csme
Mac补丁地址:
谷歌:https://drive.google.com/open?id=1GRwysPQhOYnrriZTKSRc05Mf_Q7xd5DC
百度:https://pan.baidu.com/s/1PDgXv4qrZHybyqf_niM2eA?pwd=csme
安装方法请参考上面Github地址,这边分流一个地址:https://archive.chukogals.top/posts/cos20/
结构绘制软件
/For Windows:/
Gauss View 6.0
转载自:https://github.com/Z-H-Sun/CS_CCME_Posts
百度盘:https://pan.baidu.com/share/init?surl=DWAsU46HkDF9obtxQxCOeg 密码:csme
谷歌盘:https://drive.google.com/drive/folders/18y4GHdCM6JbDAqHxMtYrAOTuGVcjoah7?usp=sharing
Gauss View 5.0(Intel Core,32bit;参考上面百度盘)
安装方法备份:https://archive.chukogals.top/posts/gvm/
Crossover+Gauss View 6.0
安装方法参考自:全网首发!如何在 M1 chip 的 macOS 上优雅的使用 Gaussian 09 与 GaussView 6(已失效,现在是webarchive的地址)
(原始地址已挂,考虑到Crossover也不贵,如果有兴趣的可以购买key,我也买了正版不过我现在没mac,有兴趣的可以找我要key用,限一个人)
安装环境:macOS Catalina 以上版本、GaussView 6
- 安装 Crossover;(按住ctrl+点击打开安装)
- 打开 Crossover,选择 安装 Windows 程序;【注意,Crossover是商业软件,请低调使用】
- 点击左下角,查看所有应用程序;
- 选择 未列出的应用程序,点击继续;
- 选择上方的安装包 —> 下载安装程序 —> 选择下载的 GV6.0.16_win64.exe,点击继续;
- 容器使用“新 Windows 10 64-bit 容器”,右边取个名字,然后按继续,就像 Windows 那样安装;
- 安装需要序列号,可以在文件夹的文本文件中找到;
- 既可通过启动区启动GV,也可也通过crossover启动。
结构查看软件
Gauss View。
另外补充CYLview(双平台):
http://www.cylview.org/download.html
相关操作暂时参考:
使用CYLview绘制高质量分子结构图和制作化学反应演示动画
看了一下,好像还可以导出优化过程中的能量变化曲线,可能有些用吧。
结构计算软件
因为我们服务器上已经配置好了Gaussian 16的linux版,因此这里不再对结构计算软件的相关内容进行说明。
具体建议参考Gaussian的安装方法及运行时的相关问题进行浏览。
M1芯片和64位MacOS系统参考上面的文章。
核心软件下载
Gaussian和Gview所有相关下载地址:
参考自:CS_CCME_Posts
另外这个项目也很适合star。
Gaussian Win64位:
文件来自上海交大网络信息中心 已经内置了激活码所以没什么好说的。
服务器帐号的申请与环境的配置
本文中全部标为/示例文字/的部分请不要复制到SSH中。
系统环境
个人用服务器 超级计算机服务器均采用Linux系统,请参考查阅Linux系统相关使用教程。
武大超算使用手册:连接超算 · GitBook
Linux的基础知识
shell
Shell简单来说是一种操控系统,或者包装应用程序的操控界面。它包括指令行界面command-line interface (CLI)【如DOS】、图形用户界面graphical user interface (GUI)等【如Windows】。Linux下有很多种shell,诸如bash、csh、ksh等,相当于不同的文本终端的界面。不同的shell下一些命令不同,比如设置环境变量,在bash里用export命令,而在csh里用set命令。其中Bash是最常见的shell,也是大多数Linux系统默认的shell。
用户主目录
Linux下每个用户都有自己的主目录。默认情况下,你以命令行方式登录系统后,你当前所在的目录就是用户主目录,可以敲pwd命令显示当前的目录。如果你是普通用户的话,那么主目录将为/home/[用户名]/。如果你是root的话,那么主目录就是/root/。
文件权限
Linux下每个文件都有称为权限的属性,可以用chmod命令来修改。权限包括是否可执行、是否可读取、是否可修改。比如chmod +x ./yourfile 就会把当前目录下的yourfile加上可执行权限,再比如chmod 750 /user/test.exe命令就会把/user/test.exe文件设为750权限。这里750的百位数、十位数、个位数分别对应这个文件对于当前用户的权限、对于与当前用户同组用户的权限、对于其它组用户的权限。7对应完整的权限,即可执行、可读取、可写入,5代表可读取、可执行,而0代表所有权限都没有。更多关于chmod的知识请自行google。
.bashrc文件
每个用户主目录下都会有.bashrc文件,这是个文本文件,里面记录了当你每次登录bash shell的终端的时候都会自动执行的命令。因此如果有些命令你希望每次登录终端时都能生效,那么把命令写入.bashrc文件即可。.bashrc文件里有一些自带的内容,那些都不用管,自己加新的命令往此文件末尾添加即可。Linux下所有开头为.的文件都是隐藏文件,直接用ls命令看不到,而得用诸如ls -a才能看到。如果你是在系统自带的图形界面的文件浏览器下,得按一次Ctrl+H才能看到隐藏文件。显然,.bashrc文件也是个隐藏文件。
高斯软件的配置
正常的高斯安装方法
参考自:Gaussian的安装方法及运行时的相关问题 - 思想家公社的门口:量子化学·分子模拟·二次元
- 假设当前用户名是a,要把Gaussian安装到自己的主目录下,且用的是bash,那么安装过程为:
- 把Gaussian压缩包解压到/home/a/目录下
- 建立一个文件夹用于储存Gaussian运行过程中产生的临时文件,位置随意。比如此例我们建立/home/a/g09/scratch。
- 用vi或者gedit等文本编辑器打开/home/a/.bashrc,在里面加入以下语句然后保存文件。
export g09root=/home/a
export GAUSS_SCRDIR=/home/a/g09/scratch
source /home/a/g09/bsd/g09.profile- 其中,g09root环境变量用来说明Gaussian目录被放在了哪个目录下。GAUSS_SCRDIR环境变量用来说明哪个文件夹作为临时文件夹。source命令用来执行Gaussian自带的脚本文件,其中包含了各种配置Gaussian运行环境的命令。
- 重新进入终端使以上内容生效(如果你是用Linux图形环境,就关闭终端窗口,然后重新打开。如果你是通过ssh方式登录服务器,就断开连接,然后重新连接服务器)
- Gaussian目录下的Default.Route用来设定默认用的计算资源(如果没有此文件就新建一个),-M-设置默认用的最大内存量(一般用MB或GB为单位),-P-设置默认用多少CPU核数来并行计算。比如我们想默认用36个核心、最大60GB内存做计算,
就在/home/a/g09/Default.Route里面写入以下内容
-M- 60GB
-P- 36- 此文件中的设置优先级低于输入文件里的%mem和%nproc设置。因此,输入文件里没写%mem的时候才会用这里的-M-设置,输入文件里没写%nproc的时候才会使用这里的-P-设置(如果你既没在这里设-P-也没写%nproc,那么程序只用单核计算。如果你既没在这里设-M-也没写%mem,那么G09最多用256MB内存,G16最多用800MB内存)。
- 最后,切换到/home/a/g09/目录,运行chmod 750 -R命令,之后Gaussian就可以使用了。这里-R代表对当前目录下所有文件和所有子目录下的文件都设置权限。
- 如果你用的是Gaussian 16,只需把上面过程中所有g09改成g16即可,其它没有任何差别。
武大超算这边的执行方法
先通过ftp将g16转移到自己的超算文件夹中,再通过以下代码进行解压和赋权:
cd g16/
tar xvfJ E6B-114X.tbJ /*转移和解压相关的软件压缩包*/
chmod 750 -R * /*将该目录增加可执行权限*/
cd bin/
chmod 750 -R *注意:我才发现组里使用的g16的版本(C01)和网上流传的版本(A03)不一样而且压缩包格式也不同,请注意一下。
任务进度提交、查看常用代码
参考自:连接超算 · GitBook
ls /*显示当前目录下的内容*/
cd /*转移到某一目录下*/
qstat -a /*显示当前提交任务的运行状态*/
如:
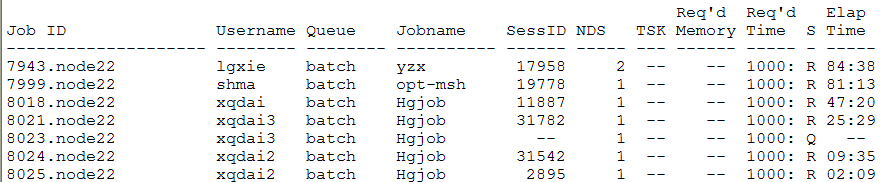
sinfo /*显示当前服务器的核心/线程占用情况*/
其中 STATE 常见的有:
- alloc,节点已经被分配给作业任务
- down,节点当前不可用
- drain,节点被管理员设置为退出服务
- idle,节点当前空闲
- mix,节点有部分计算资源已被分配,还有部分资源空闲可用
squeue /*显示当前提交任务的排队情况*/
显示的信息包括以下内容:
- JobID,作业编号
- PARTITION,作业在哪个分区上运行
- NAME,作业名称,默认是作业脚本的名字
- USER,作业的所有者
ST,作业当前状态,详见 Job State Codes ,常见的有
- CG 作业正在完成
- F 作业失败
- PD 作业正在等待分配资源
- R 作业正在运行
- TIME,作业已运行时间
- NODES,作业占用的计算节点数
- NODELIST,作业占用的计算节点名
CPU脚本示例:
#!/bin/bash
#SBATCH --account=admin——teacher
#SBATCH --partition=hpib
#SBATCH --nodes=1
#SBATCH --ntasks=16命名为myjob.sh,然后 sbatch myjob.sh 即可
常见错误排除
我在配置环境中,曾遇到无法正常输出文件的情况。
经检查,发现问题出自使用的脚本中,没有正确指定高斯临时文件的位置。
Before:
output.write(“GAUSS_SCRDIR=/scratch/myname/gaussian16_scrdir\n”)
After:
output.write(“GAUSS_SCRDIR=/scratch/myname\n”)
前者是在自用服务器copy来的脚本,可见脚本之间并不存在完全通用的情况。
Gauss View使用基础
相关教材推荐
南开大学的
中国科大超级计算中心的
GaussView程序介绍
阿部穣里老师的课件:量子化学演習 2020オンライン
(备份:https://disk.yandex.com/d/locju4Q3clPaow)
倒是很详细的说了GaussView的使用方法和调用Gw16的相关内容,直接打开【GaussView と Gaussian の基本的な使い方】这篇PDF看看就行。
相关操作要点:
- .gjf文件
- 原子的增减与替换
- 添加固定模版结构
- 结构参数的微调
- 结构状态(多重态)的调整方法
- 查看循环次数/优化过程
- 错误信息的排查
软件进阶?
输入文件的处理
正常的输入文件是无法直接投到高斯工作的,需要通过修改文件进行调整。
未经调整的文件:
%chk=root\sample.chk
# hf/3-21g geom=connectivity
Title Card Required
0 1
C A B C
H A B C
1 2 1.0 3 1.0 4 1.0 5 1.0
2
3
4
5
_
_(此处大多需要预留两行,用下划线代替)调整后的文件:
%mem=60000MB
%nprocshared=16
# opt freq b3lyp/6-31g(d) 5d empiricaldispersion=gd3bj
Title Card Required
0 1
C A B C
H A B C
_
_(此处大多需要预留两行,用下划线代替)输入文件的固定开头(link 0 command)
Control files&system resources
%chk=h2o # 链接0部分
%mem=64GB
%nproc=26主要设置运行该计算时需要调用的系统参数。
上面的后两排最为常用。
%mem=N
设置用于作业的动态内存(RAM)调用量。默认值为6MW。N后面可以有一个单位名称:KB、MB、GB、KW、MW或GW。
%nproc(l)=N or %nprocshared=N
设置作业最多可使用的处理器个数(核心)上限。
%chk=file
查找检查点文件(.chk)并命名
%rwf=file
查找并命名单个统一的.rwf文件
%KJob LN [M]
告诉程序第M次发生N号序列程序后停止运行
%save
在Link0在运行结束时保存临时文件
%subst LN dir
给Link0来自备用目录的一个可执行文件链接
计算模型与计算方法的选择
在固定开头的下方一般有以下三个部分:
#p hf/sto-3g opt /*计算模型的选择(执行路径部分)*/
water optimization /*计算类型*/
Title Card Required
0 1 /*价态与多重态(分子说明部分)*/
C A B C
H A B C /*坐标*/
O A B C
_
_(此处大多需要预留两行,用下划线代替)关于其中内容的解读和基组选择等内容,可以参考课题笔记1中的大量介绍内容。
输出文件的理解
(待更新)